Berkeley Drosophila Genome Project
BDGP Resources
Gene Disruptions Using P Transposable Elements
PNAS 92:10824-10830, 1995 Allan C. Spradling#, Dianne Stern# , Istvan Kiss&, John Roote*, Todd Laverty+, and Gerald M. Rubin+ # Howard Hughes Medical Institute Research Laboratories, Carnegie Institution of Washington, 115 W. University Parkway, Baltimore, MD 21210 Institute of Genetics, Biological Research Center, Hungarian Academy of Sciences, H-6701 Szeged, Hungary * Department of Genetics, Cambridge University, Downing St., Cambridge, CB2 3EH England
- Howard Hughes Medical Institute Research Laboratories, Department of Molecular & Cell Biology, University of California at Berkeley, Berkeley, CA 94720-3200
Biologists require genetic as well as molecular tools to decipher genomic information and ultimately to understand gene function. The Berkeley Drosophila Genome Project is addressing these needs with a massive gene disruption project that uses individual, genetically engineered P transposable elements to target open reading frames throughout the Drosophila genome. DNA flanking the insertions is sequenced, thereby placing an extensive series of genetic markers on the physical genomic map and associating insertions with specific open reading frames and genes. Insertions from the collection now lie within or near most Drosophila genes, greatly reducing the time required to identify new mutations and analyze gene functions. Information revealed these studies reveal about P element site specificity is being used to target the remaining open reading frames. Genetics provides the most powerful approach available to understand the function of each human gene and to decipher the role played by the large noncoding component of the human genome. Model organisms, such as Drosophila melanogaster, share many genes with humans whose sequences and functions have been conserved. In addition to myriad similarities in cellular structure and function, humans and Drosophila share pathways for intercellular signaling (1), developmental patterning (2), learning and behavior (3), as well as tumor formation and metastasis (4). The fruit fly provides a powerful system to study the function of conserved genes since, unlike humans, any open reading frame (ORF) within the fruit fly genome can be mutated and subjected to detailed functional analysis within the context of an intact organism. The Berkeley Drosophila Genome Project (BDGP) has as its primary goal to map and sequence the 120 Mb of DNA comprising the euchromatic-i.e., nonrepetitive-regions of the four Drosophila chromosomes (see for examples ref. 5; W. Kimmerly, K. Stultz, K. Lewis, V. Lustre, S. Lewis, D. Sun, R. Romero, C. Martin, and M. Palazzolo, personal communication). Novel informatics tools are being developed to collect, analyze and distribute these data. If this vast new store of structural information is to enhance our understanding of human biology, however, it must be accompanied by more efficient methods to correlate genes with functions. Currently, database searches identify similarities to a given query sequence more than a third of the time. Moreover, these similarities are often associated with biological meaning because many of the entries in the public database have been deposited by individuals involved in hypothesis driven research. With the onset of sequencing whole genomes and large cDNA collections, database similarities will be found more frequently. However, because future database submissions will come primarily from genome-sequencing efforts which select targets based on developing complete databases rather than on their biological function, the frequency of association of a query sequence with well-characterized biochemical mechanisms is unlikely to rise in parallel. If the model organism genome projects are to be maximally useful in assigning function to human DNA sequences, they must be accompanied by genetic studies so that not only the sequences of the genes, but also their biological functions, are determined. To facilitate that end, BDGP has adopted a broad approach that combines the determination of the genomic sequence with the development and application of methods for large-scale functional analysis. In the past, the effort required to disrupt a particular gene or to identify the ORF responsible for a mutant strain's interesting properties has varied widely and has frequently slowed progress. Consequently, BDGP has undertaken a gene-disruption project to address this rate-limiting step in utilizing Drosophila to assign function to human genes. The BDGP gene disruption project consists of a large collection of Drosophila strains that each contain a single genetically engineered P transposable element inserted in a defined genomic region. The BDGP strain library, which is freely available from the Bloomington, IN, Drosophila stock center, greatly facilitates both gene disruption and mutant identification. Investigators interested in a particular sequenced ORF can often find a strain from the collection in which that ORF is insertionally mutated. Otherwise insertions can usually be obtained close enough to the ORF of interest to rapidly generate the desired mutations. In addition, BDGP strains make it much easier to associate the thousands of genes Drosophila geneticists have defined over the last several decades with specific transcription units and their protein products. BDGP lines can be identified that either disrupt such genes or otherwise delimit their positions on the physical map to small molecular intervals. Finally, the inserted P elements in BDGP lines carry enhancer-traps (6) that can be used to efficiently acquire information about the expression pattern of disrupted genes. The strains in the current collection disrupt 20 - 25 % of essential genes, provide information on their expression patterns, and link the genetic, cytogenetic, and physical maps of the Drosophila genome at 100-kb intervals. The approaches made possible by the BDGP strains are reducing or eliminating long delays in obtaining the tools needed to study gene function. In this report we discuss the current status of the BDGP gene disruption library and report some of the results revealed by this project. Origin of the BDGP Gene-Disruption Project Transposable elements provide a potent means of correlating genetic and molecular information because they generate a simple, reproducible lesion upon insertion that can be detected much more easily than damage produced by other mutagens (7,8). In D. melanogaster the P transposable element has been particularly useful because it moves with high frequency but can be controlled by limiting the availability of an element-encoded transposase (9,10). Early efforts focused on cloning specific genes by mobilizing large numbers of natural P elements (reviewed in ref. 11). The resulting genetic lines were unsuitable for genetic or phenotypic studies without extensive outcrossing to remove extraneous P elements and were rarely saved once flanking DNA had been cloned. P element-mediated transformation allowed strains containing just one or a few elements to be constructed (12). However, only a limited number of strains could be generated by microinjecting DNA, and the insertions could not be targeted into genes of particular interest. In 1988 Cooley et al. (13) showed that individual, experimentally modified P elements can be mobilized in large genetic screens to generate thousands of stable mutant strains. About 15% of such transposon insertions disrupted a gene required for viability or fertility, while the remaining insertions presumably integrated into phenotypically silent genes or spacer regions. They proposed that single-insert stocks associated with mutations be collected and used to select a minimal subset of strains (an "insertion library") to maintain in a Stock Center as a community resource with diverse applications in genomic analysis. By identifying mutations that disrupted different genes from throughout the genome for the library, the inability to target P elements into particular sites would be largely overcome. The size of the library presented an obstacle; collections of Drosophila mutant stocks must remain small since strains cannot be reliably maintained in a frozen state. This problem could be minimized, however, by eliminating redundant or aberrant lines so that the collection would not grow beyond the number of mutable genes and thus remain small enough to maintain and distribute widely. The prospect of a P element insertion library was further enhanced when P element constructs that could be used for gene disruption were engineered to facilitate the cloning of flanking genomic DNA (14), the detection of gene expression patterns (6), the mis-expression of genes in developmental patterns (15) and the mediation of site-specific recombination (16). Numerous mutant strains containing single P element insertions were subsequently generated in several laboratories (refs. 17-21; M. Scott and M. Fuller, personal communication). However, it remained impractical for any single group to determine all their insertion sites and genetic properties. Without this information, an insertion library of maximum utility and feasible size could not be created, and the long-term maintenance of these strains remained in doubt. Recognizing that for a model organism, genetic tools are highly synergistic with molecular and informatics tools, BDGP undertook to characterize all available single P element insertion lines and to use them to generate a comprehensive gene-disruption library. As a byproduct, it was realized that the strains would prove extremely useful for linking the physical and genetic maps. When the project began in 1993, more than 3,000 strains in which single P element insertions had been associated with recessive lethal or sterile phenotypes were collected from six laboratories (Table 1). The gene-disruption library was assembled from this starting material, and it continues to be expanded today. In their starting state, the lines were of limited use to the research community and too numerous to be maintained in stock centers. Since most of the transposon insertion sites were unknown, investigators would have to study thousands of lines instead of just the few whose P element insertions that lay in a genomic region of interest. Furthermore, many of strains in the collection mutated the same genes, due to the existence of P element insertion hotspots. Others contained background mutations that were responsible for the mutant phenotype instead of the inserted transposon.
Table 1. Sources of P element lines
| Originating Laboratory |
Number of Strains
|
Insertions on II
|
Insertions on III
|
Reference |
| A. C. S. | 1,124 | 460 | 664 | 13, 18 |
| G.M. R. | 146 | 50 | 96 | 19 |
| L. and Y.-N. Jan | 201 | 0 | 201 | 17 |
| M. Scott and M. Fuller | 114 | 35 | 79 | Personal communication |
| I. K* | 1,500 | 1,500 | 0 | 20 |
| A. Laughnon | 99 | 0 | 99 | 21 |
| Total | 3,184 | 2,045 | 1,139 |
*Estimated number of independently derived lines with < 3 insertions
High-Resolution Cytogenetic Mapping of Transposon Insertion Sites Accurately localizing the sites of transposon insertion in all the starting strains was the key to constructing a nonredundant library. In situ hybridization to salivary gland polytene chromosomes can physically map a given DNA sequence with an accuracy of 1-2 polytene bands (Fig. 1). The 4015 bands recognized on the major autosomes are divided into 485 polytene subdivisions (21A-100F), an average of 8 bands per subdivision. On the basis of microspectrophotometry and comparisons with chromosomal walks, an average band contains 25 kb of genomic DNA. Thus, if carried out with maximal accuracy, the insertions would provide valuable guideposts that could be used throughout the genome. So far the insertions in 2785 of the lines have been mapped by in situ hybridization (Fig. 1). These sites are distributed throughout the autosomes in a pattern indistinguishable from random (once the effects of a small number of P element insertion hotspots are removed). For example, 410 of 470 euchromatic subdivisions contain at least one insertion. At least 1200 different autosomal insertion sites have been resolved; hence, the average distance between elements within autosomal regions is about 85 kb.
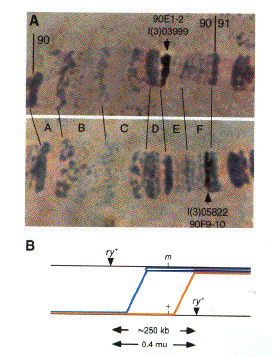 Fig. 1. Localization of
insertions to precise genomic regions and their use in fine-scale recombination mapping. (A) The insertions
in line l(2)03999 and l(2)05822 were mapped to bands 90E1-2 and 90F9-10, respectively, by polytene chromosome in
situ hybridization using an insertion-specific probe. Pictures of the hybridization signals similar to those
shown were recorded with a video camera, digitized, and stored in the BDGP database. (B) Recombinants
between the two insertion sites, which each carry a functional rosy+ (ry+) eye-color gene, were recovered
among the progeny of heterozygous females as flies with rosy mutant eye color. The location of two such crossovers is
drawn relative to the position of an arbitrary mutation (m) located within the interval defined by the
P element insertions. The inheritance of the mutation itself or molecular polymorphisms within the interval
among the recombinants can be used to assist in positionally cloning the mutated gene by more precisely determining
its location. The blue line shows the position of a hypothetical recombination event that would produce a ry-
m- chromosome, while the recombination event illustrated by the yellow line would produce a ry- m+
chromosome. Recombinants can be recognized by their unique ry- eye color, making it possible to readily
isolate 50 -100 independent recombinant chromosomes. The relative number of these two classes of recombinants gives
an estimate of the location of the mutant within the interval between the P element insertions. Since the
interval is itself only 250 kb, it is generally possible to map a mutation to a 20- to 30-kb region in this way.
Further refinement and conformation can be provided by assaying the recombinant chromosomes for the presence of
physically mapped molecular polymorphisms (22).
Fig. 1. Localization of
insertions to precise genomic regions and their use in fine-scale recombination mapping. (A) The insertions
in line l(2)03999 and l(2)05822 were mapped to bands 90E1-2 and 90F9-10, respectively, by polytene chromosome in
situ hybridization using an insertion-specific probe. Pictures of the hybridization signals similar to those
shown were recorded with a video camera, digitized, and stored in the BDGP database. (B) Recombinants
between the two insertion sites, which each carry a functional rosy+ (ry+) eye-color gene, were recovered
among the progeny of heterozygous females as flies with rosy mutant eye color. The location of two such crossovers is
drawn relative to the position of an arbitrary mutation (m) located within the interval defined by the
P element insertions. The inheritance of the mutation itself or molecular polymorphisms within the interval
among the recombinants can be used to assist in positionally cloning the mutated gene by more precisely determining
its location. The blue line shows the position of a hypothetical recombination event that would produce a ry-
m- chromosome, while the recombination event illustrated by the yellow line would produce a ry- m+
chromosome. Recombinants can be recognized by their unique ry- eye color, making it possible to readily
isolate 50 -100 independent recombinant chromosomes. The relative number of these two classes of recombinants gives
an estimate of the location of the mutant within the interval between the P element insertions. Since the
interval is itself only 250 kb, it is generally possible to map a mutation to a 20- to 30-kb region in this way.
Further refinement and conformation can be provided by assaying the recombinant chromosomes for the presence of
physically mapped molecular polymorphisms (22).
We took advantage of the fact that many genes had independently been mutated in more than one line to determine how accurately the insertions had been localized. Since most Drosophila genes studied to date are smaller than 50 kb, one would expect that the insertions in allelic lines would map within the same or an adjacent polytene band. Errors in localization would show up as allelic lines whose insertions had been mapped at more widely separated sites. Complementation crosses identified 750 lines that fell into 180 groups with between two and 25 alleles. The average standard deviation in the map position of allelic insertions from this sample was only 0.61 +/- 0.69 band. Since these tests would have revealed mistakes in localization as large as 8-16 polytene chromosome bands, their reproducibility was undprecedented. Pictures of the hybridization signals from each line were digitally recorded and have been distributed as part of the BDGP database (see Figure 2). Thus, specific lines from the collection can be requested whose insertions have been shown to reside in virtually any genomic interval.
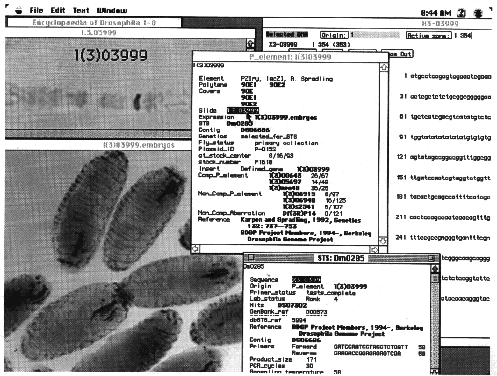 Fig. 2. Data from the
gene disruption project as displayed by the Encyclopædia of Drosophila database browser. The Encyclopædia of
Drosophila is a joint product of the BDGP and FlyBase (23). It is implemented in a version of ACeDB, a genome
database program written for the UNIX operating systems by Richard Durbin (Sanger Center, Cambs, U.K.) and Jean
Thierry-Mieg (Centre National de la Recherche Scientifique, Monpellier, France). The original customization of ACeDB
for use with Drosophila was done by Suzanna Lewis (BDGP) to produce Flydb, the laboratory database of the
BDGP and further enhancements for the Encyclopædia of Drosophila were made by Suzanna Lewis and Cyrus Harmon (BDGP).
A Macintosh-compatible version of ACeDB was written by Frank Eeckman (Lawrence Berkeley Laboratory), Richard Durbin
and Cyrus Harmon and was further customized for the Encyclopædia of Drosophila by Cyrus Harmon. The Encyclopædia of
Drosophila displays published and unpublished data of the BDGP, as well as data collected by FlyBase from the
scientific literature and other genome projects. The individual windows illustrate the organization of information
relevant to each strain in the BDGP gene-disruption library, using strain l(3)03999 as an example. The window
entitled P_element: l(3)03999 summarizes general information, such as the structure of the inserted element, the
polytene chromosome location of the insertion, the name of the squence-tagged site (STS) derived from the insertion,
the name of the contig in which this STS lies, the date the line was sent to the Stock Center, and the results of
genetic complementation tests with other P element insertions and chromosomal deletions. Double clicking on
individual items in this window opens other windows that display more detailed information. For example, selecting
the slide number, l.3.03999, opens a window that shows a digitized image of the chromosomal in situ
hybridization used to map the insertion. Double clicking in the expression field opens a window, entitled
l(3)03999.embryos, that displays the embryonic expression pattern of the enhancer trap associated with this
insertion. More information about the STS derived from this insertion can be obtained by opening the window entitled
STS: Dm0285. From within this window, the window entitled X3-03999, which contains the sequence of the STS, can be
opened.
Fig. 2. Data from the
gene disruption project as displayed by the Encyclopædia of Drosophila database browser. The Encyclopædia of
Drosophila is a joint product of the BDGP and FlyBase (23). It is implemented in a version of ACeDB, a genome
database program written for the UNIX operating systems by Richard Durbin (Sanger Center, Cambs, U.K.) and Jean
Thierry-Mieg (Centre National de la Recherche Scientifique, Monpellier, France). The original customization of ACeDB
for use with Drosophila was done by Suzanna Lewis (BDGP) to produce Flydb, the laboratory database of the
BDGP and further enhancements for the Encyclopædia of Drosophila were made by Suzanna Lewis and Cyrus Harmon (BDGP).
A Macintosh-compatible version of ACeDB was written by Frank Eeckman (Lawrence Berkeley Laboratory), Richard Durbin
and Cyrus Harmon and was further customized for the Encyclopædia of Drosophila by Cyrus Harmon. The Encyclopædia of
Drosophila displays published and unpublished data of the BDGP, as well as data collected by FlyBase from the
scientific literature and other genome projects. The individual windows illustrate the organization of information
relevant to each strain in the BDGP gene-disruption library, using strain l(3)03999 as an example. The window
entitled P_element: l(3)03999 summarizes general information, such as the structure of the inserted element, the
polytene chromosome location of the insertion, the name of the squence-tagged site (STS) derived from the insertion,
the name of the contig in which this STS lies, the date the line was sent to the Stock Center, and the results of
genetic complementation tests with other P element insertions and chromosomal deletions. Double clicking on
individual items in this window opens other windows that display more detailed information. For example, selecting
the slide number, l.3.03999, opens a window that shows a digitized image of the chromosomal in situ
hybridization used to map the insertion. Double clicking in the expression field opens a window, entitled
l(3)03999.embryos, that displays the embryonic expression pattern of the enhancer trap associated with this
insertion. More information about the STS derived from this insertion can be obtained by opening the window entitled
STS: Dm0285. From within this window, the window entitled X3-03999, which contains the sequence of the STS, can be
opened.
Disrupting Genes and Mapping Mutations on the Basis of Their Location Simply knowing the sites of transposon insertion to high accuracy makes many types of genetic analysis possible when using lines from the BDGP library. Mutations are frequently desired in an ORF that was identified and cloned by a nonredundant library. Once the cytogenetic location of any gene has been determined by in situ hybridization, lines from the BDGP library can be obtained whose insertions are located nearby. These transposons contain an easily scored eye-color marker and can be remobilized efficiently. Consequently, the inherent tendency of P elements to transpose "locally" (within about 100 kb) can be utilized to preferentially mutate the region containing the gene of interest (24). Moreover, by selecting for loss of the marker gene following re-mobilization or x-ray treatment, small chromosomal deletions in the surrounding region can be generated, some of which are likely to remove the desired locus. Frequently, mutations disrupting a biological process of interest have been identified in chemical mutagenesis screens, but their locations are not known accurately enough for efficient cloning. Using BDGP lines, any mutation can be accurately mapped by recombination relative to nearby insertions (see Fig. 1). This allows the mutation to be placed within specific physical intervals defined by their nearest two flanking insertions. Analysis of recombinants between parental insert-bearing chromosomes, in particular the number of recombinants seen between the mutation and each P element, allows the approximate position of the mutation within the interval to be determined. Moreover, the resulting recombinant chromosomes can be scored for DNA sequence polymorphisms within the interval to further refine the position of the mutation (for example, see ref. 22). The two insertions shown in Fig. 1 are separated by approximately 250 kb. From 23,000 progeny a total of 46 recombinants were selected in which crossing-over had occurred between the two elements simply by selecting files that had lost both eye-color markers. This provides an average of about one crossover every 5 kb, which in most genomic regions would be sufficient to map a point mutation to the site of one or a very few specific transcription units. The correct transcript can subsequently be identified by testing the ability of various DNA segments from the region to complement the mutant defect in transgenic flies or by comparing the sequence of mutant and wild type alleles. Identifying Disrupted Genes As the size of the BDGP gene disruption library grows, the density of insertions along the physical map will increase, and the with it the power and precision of the local mutagenesis and recombination mapping methods described above. However, the fraction of Drosophila genes that have been directly disrupted by a transposon insertion in the collection will also grow and will reduce the need for these methods. Strains whose insertions directly disrupt genes of interest are ideal tools to expedite studies of gene function. For example, it is usually possible to identify rapidly the transcription unit an insertion has disrupted by looking in the mutant for altered transcripts near the insertion site. Unlike mutations generated by chemical mutagens or radiation, single P element insertions allow new alleles of the gene to be generated rapidly by imprecisely excising the original element. Studying a range of mutant alleles that includes true nulls is frequently important for understanding gene function. Imprecise excisions can be selected that delete the gene's promoter and coding sequences, revealing its true "null" phenotype. Because of the importance of direct gene disruptions, a major goal of the BDGP is to identify as many lines as possible from the starting collection whose insertions disrupt different genes that cause scorable phenotypes. This requires proving that the mutant phenotype originally associated with the line is in fact caused by the transposable element insertion, rather than a secondary lesion that arose during the screen. Moreover, the mutation must define a previously undisrupted genetic locus and not simply represent another allele of a gene already disrupted within an existing strain in the library. Stains with these properties become a permanent part of the final library. The number of different genes that are ultimately represented should be limited only by any inherent site specificity that restricts the ability of P elements to mutate some loci and by the number of starting strains that can be generated and analyzed. Two methods are being used to verify that the P element insertion is indeed the cause of the lethal mutation and thereby increase the size of the final gene disruption library. The first is to identify loci with two or more independent P insertion alleles. Control crosses using the starting lines show there is a negligible probability that two lines will fail to complement for reasons unrelated to their cytogenetically similar P insertions. The crosses described above to test the accuracy of insert localization have so far identified 180 loci with multiple alleles. A second and more generally applicable method is to determine if the mutant phenotype associated with each starting strain is uncovered when the region surrounding the insertion site is removed by a deletion. Small deletions are available that allow such tests to be carried out on the majority of the starting strains (see Fig. 3). An additional advantage of this method is that lines bearing background mutations can be positively identified, and the affected lines discarded. New deletions are being constructed to allow insertions in the remaining genomic regions to be tested by this method.
 Figure 3. Complementation
data illustrating the use of chromosomal deletions to verify insertion lines. Genetic complementation results from
polytene subdivision 63F-64D are summarized below a picture of the polytene banding pattern of this region. The
chromosome region between bands 63F4-7 and 64C13-15, which polytene cytology shows to be missing in strain
Df(3L)GN24, is indicated by a black box. Below the box are the names of seven strains associated with lethal
mutations from the starting collection whose insertions were mapped to this region by in situ hybridization.
An X indicates that the strain failed to complement the deletion, while an O indicates that the lethal mutation
survives in combination with Df(3L)GN24. The perfect correlation between the deletion endpoints and
complementation behavior indicates that the lethal mutations in the five lines lying inside Df(3L)GN24 are
caused by their P element insertions. One line, l(3)02333 (not shown), contained an insertion at 64B16-17
but complemented Df(3L)GN24 and was therefore discarded.
Figure 3. Complementation
data illustrating the use of chromosomal deletions to verify insertion lines. Genetic complementation results from
polytene subdivision 63F-64D are summarized below a picture of the polytene banding pattern of this region. The
chromosome region between bands 63F4-7 and 64C13-15, which polytene cytology shows to be missing in strain
Df(3L)GN24, is indicated by a black box. Below the box are the names of seven strains associated with lethal
mutations from the starting collection whose insertions were mapped to this region by in situ hybridization.
An X indicates that the strain failed to complement the deletion, while an O indicates that the lethal mutation
survives in combination with Df(3L)GN24. The perfect correlation between the deletion endpoints and
complementation behavior indicates that the lethal mutations in the five lines lying inside Df(3L)GN24 are
caused by their P element insertions. One line, l(3)02333 (not shown), contained an insertion at 64B16-17
but complemented Df(3L)GN24 and was therefore discarded.
At present, slightly more than 700 lines have been incorporated into the disruption library, representing 20% of the estimated 3500 autosomal genes that mutate to a scorable phenotype. All these lines have been deposited for pubic distribution at the Drosophila Stock Center in Bloomington, IN. Strains from the BDGP collection already rank among the most frequently requested items and accounted for more than 30% of all strains shipped during 1994, their first full year of availability. The molecular structures of the genes disrupted in the library are being learned from two major sources. First, a flanking sequence tag is generated for each insertion in order to localize it precisely on the genomic sequence, to generate an STS for the BDGP physical mapping project, and to assist in gene identification. Generating the sequence tags is greatly facilitated by the presence of internal cloning sequences in the inserted transposons that allow 5' flanking DNA to be cloned by plasmid rescue (12). Subsequently, about 350 bp of 5' sequence is determined and compared to DNA sequence libraries to learn if the insertion falls within a previously sequenced region. Additionally, the flanking sequence is translated in all six reading frames and compared with proteins from all organisms. The results of analyzing the first 280 flanking sequence tags showed that approximately 20% of were homologous to Drosophila sequences previously reported in public databases, leading in most cases to the molecular identification of the disrupted gene. Nearly half of these disrupt genes encoding proteins with extensive similarity to known human genes. Of course, these percentages underestimate the true fraction of insertions that disrupt known Drosophila genes and genes with mammalian relatives because they are based on such a short sequence of genomic DNA and because the sequence of most human genes is not yet available for comparison. The second approach to gene identification is the Drosophila research community, which has a close working relationship with BDGP. The chromosomal insertion site data and genetic complementation results from each line are widely distributed via the Internet as part of the FlyBase (23) database project and in the form of an integrated genome browser known as "Encyclopædia of Drosophila" that is produced in a collaborative effort by the BDGP and FlyBase and distributed by CD-ROM (Fig. 2). Information on the "enhancer trap" expression patterns in these lines is also being collected and incorporated into the database. These patterns are known to frequently reflect the expression pattern of the mutated gene, so this information allows genes to be selected for further study on the basis of their probable patterns of expression in diverse tissues. With this information, the BDGP insertion lines located near genes currently undergoing study by members of the research community are routinely requested from the Stock Center and molecularly characterized, and the results reported. Estimating Mutational Saturation by Using a 1.8-Mb Genomic Region The genomic region surrounding the Alcohol dehydrogenase (Adh) gene in polytene divisions 34D-36A has been extensively studied (see ref. 25). Forty eight complementation groups that mutate to lethality have been defined from mutagenesis screens that appear to have saturated mutable vital loci. We selected this region to estimate the final saturation level of autosomal loci that could be expected using the starting strains. All lines failing to complement deficiencies known to uncover the 34D to 36A region were identified and crossed to representative complementation groups defining the 48 known vital loci. In addition, the cytogenetic locations of the insertions in these lines were determined to ensure that the mutations were associated with a P element insertion. The results from this study were highly informative (Fig. 4). A total of 16 of the 48 vital genes (33%) were mutated by lines in the collection. Thus, if the 34D-36A region is representative, 1/3 of all vital second chromosome genes ( 500 genes) will have been mutated by verified single-insert lines when the analysis of the 3,000 starting strains is complete. A slightly lower number of third chromosome genes is predicted (400) because fewer starting insertions were recovered on that chromosome. These results are also consistent with our previous estimates of inter-insert distances. Since the 34D-36A region is estimated contain 1.8 Mb of DNA, the average distance between insertions mutating vital genes would be approximately 113 kb.
Figure 4. Diagram of the vital genetic loci in the chromosomal region 34D-36A. This region contains approximately 1.8 Mb of DNA. The solid boxes represent the polytene chromosome bands of the cytogenetic map. The names and approximate positions of genetic loci are indicated (J.R. and M. Ashburner, unpublished data). The 48 loci can mutate to lethality, over one-third (16 / 48; 33%) of the previously known vital complementation groups in this region were mutated by P element insertion strains in the current collection. These genes are highlighted.
While not detracting from the usefulness of the collection, this is a lower value than would have been expected if P elements could inactivate any gene. The total number of independent second chromosome lines is approximately 2000, while 1600 lethal complementation groups are estimated to reside on this chromosome. If they were equally mutable with P elements more than 67% of the loci (rather than 33%) would contain one or more mutations. On the basis of the success rates in screens that mobilized small natural elements, P elements were previously estimated to mutate about 50% of all genes, based on the success rates in screens that mobilized small natural elements (27). It may be possible to disrupt the remaining genes by mobilizing P elements containing sequences that modify their insertional specificity or by employing other transposable elements whose movement can be controlled. The two strongest candidates for alternative elements are hobo, an inverted repeat element in the Ac family (27) and Minos, an element in the mariner/Tc family (28). The feasibility of these approaches is currently being tested. P Elements Preferentially Mutate 5' Gene Regions The information gathered on insertions from the library made it possible to examine whether P elements exhibit site specificity within genes as well. An apparent preference for insertion near gene 5' ends had been noted previously for several specific genes (29,30), but exceptions were also known. Fig. 5 shows the locations of 56 insertions present in starting lines (representing 49 different genes) whose positions have been determined at the sequence level. Insertions are plotted for comparison relative to a "generic" gene having one intron prior to the coding sequences and one intron within coding sequences. The insertions studied exhibit a strong preference for 5' untranslated regions. The next most common integration sites are located 100-200 bp upstream of the site where transcription initiates. Nine of the genes are frequent targets of P insertion and represent insertion "hotspots" (Fig. 5, filled triangles). The mapped insertions in all but one hotspot are located in transcribed regions upstream from the initiation AUG codon. Even the few insertions that are found in introns are clustered toward the 5' end, usually in introns within 5' untranslated regions or else the first intron to disrupt coding sequences. The four coding sequence insertions are located within the first 80 amino acids from the N terminus, so the 5' preference applies to all types of gene regions.
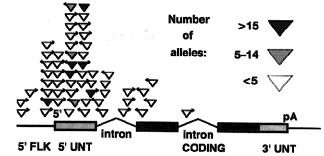 Figure 5. Preferential
P element insertion near the 5' end of transcription units. The insertion site of 56 different mutagenic
P elements whose insertion sites have been determined at the nucleotide sequence level are shown. Each
insertion site is plotted with respect to a simplified "standard" gene containing one intron before and one intron
after the AUG initiation site. The gene regions shown are as follows: 5' flanking sequences, 5' FLK (line); 5'
untranslated region, 5' UNT (hatched box); coding region (solid box); 3' untranslated region, 3' UNT (hatched box);
pA, poly(A) addition signal. The position of each insertion was plotted proportionally within the gene region shown.
Upstream insertions were plotted by assuming the 5' flanking region shown was 500 bp.
Figure 5. Preferential
P element insertion near the 5' end of transcription units. The insertion site of 56 different mutagenic
P elements whose insertion sites have been determined at the nucleotide sequence level are shown. Each
insertion site is plotted with respect to a simplified "standard" gene containing one intron before and one intron
after the AUG initiation site. The gene regions shown are as follows: 5' flanking sequences, 5' FLK (line); 5'
untranslated region, 5' UNT (hatched box); coding region (solid box); 3' untranslated region, 3' UNT (hatched box);
pA, poly(A) addition signal. The position of each insertion was plotted proportionally within the gene region shown.
Upstream insertions were plotted by assuming the 5' flanking region shown was 500 bp.
These observations strongly support previous anecdotal evidence that P elements favor 5' gene regions for insertion. Preferential integration into 5' non-coding regions is also observed with the yeast Ty1 transposon and other retroelements in studies analyzing inactivated genes (31). Host proteins, including specific transcription factors with binding sites near transcription start sites, are necessary for this specificity (32-33). Regardless of biological mechanisms underlying the tendency of the P element to insert close to the 5' end of the transcription units, the strength of this preference greatly aides in identifying the transcription unit affected by a P element-induced mutation. Disrupting "Silent " Genes The number of transcripts appears to exceed the number of mutable loci in several extensively analyzed segments of the Drosophila genome (34-35). In yeast and Caenorhabditis elegans, the majority of ORFs revealed by genomic sequencing have not been associated with mutant phenotypes, even in genomic regions that have been subjected to saturation mutagenesis (36-37). The effects of disrupting the "silent" genes revealed in these studies may be masked by the activity of related genes, or because their mutations produce defects that are not readily apparent in casual observation under normal laboratory conditions. To understand the function of such silent genes, it will be necessary to obtain null mutations, just as in the case of genes causing obvious mutant phenotypes. In many cases a phenotype can in fact be detected using more specialized assays - e.g., measuring circadian rhythms (38). However, new approaches will be required since mutations in silent genes are unrecognized in most mutant screens. Insertional mutagenesis with a single P element provides a powerful approach to obtaining mutations in silent genes when carried out in the context of whole genome sequencing. Silent genes appear to be similar in structure and regulation to vital genes. Consequently, many P element insertion lines lacking any obvious phenotype probably contain silent gene mutations. It is likely that information on the expression pattern of silent genes provided by analysis of enhancer trap insertions will be useful in guiding the choice of specialized phenotypic assays to perform on individual insertion lines. The identity of the ORF disrupted by a silent insertion would be assigned on a tentative basis entirely by molecular data. In several cases, P elements in phenotypically silent lines have been shown to disrupt genes by integration into their 5' flanking regions (A.C.S. and G.M.R., unpublished data). The frequent proximity of the insertions to the 5' portion of the transcription unit means that the gene disruption in many lines could be molecularly identified by comparing the DNA sequence flanking the insertion to genomic sequence data. Final confirmation would depend on studies of the effects of the mutation of the expression of gene products at the levels of RNA and protein. Broadening Our Concept of Genomics Multicellular organisms are proving to have far more in common at the biological level than previously suspected. Research on the most tractable models, such as Drosophila, is greatly advancing our understanding of what specific genes do, including many directly relevant to human biology and medicine. As summarized in this article, the scientists of the BDGP believes that the benefits of the whole genome analysis can best be realized if it is coupled with powerful genetic tools. We remain confident that resources, such as the gene disruption library, which integrate molecular and genetic methods will play an increasingly important role in future biological research. Acknowledgements: We thank G. Helt for carrying out database similarity searches; M. Ashburner for advice and support; D. Nakahara, S. Sundby, W.Yu and G. Doughty for help with in situ hybridization mapping of insertion lines and A. Beaton for help with genetic anaylsis. N. Patel provided images of enhancer trap expression patterns, and H. Chang provided the recombination data used in Fig. 1. We thank A. Fire, D. Koshland, M. Palazzolo, and D. Van Vactor for comments on the manuscript. The BDGP is a consortium of the Drosophila Genome Center (funded by the National Center for Human Genome Research Grant P50-HG00750; G.M.R., Principal Investigator), the Lawrence Berkeley Laboratory Human Genome Center (funded by the Department of Energy; Mohan Narla, Acting Director), and the Howard Hughes Medical Institute (through its support of the work in the G.M.R. and A.C.S. laboratories). This work was supported by the National Institutes of Health Grants P50-HG00750 and R03-HG01028. A.C.S. and G.M.R. are Investigators of the Howard Hughes Medical Institute. J.R. was supported by a Medical Research Programme Grant to M. Ashburner and D. Gubb.
REFERENCES
- Pawson, T. & Bernstein, A. (1990) Trends Genet. 6, 350-356.
- Krumlauf, R. (1992) Bioessays 14, 245-252.
- Kandel, E. & Abel, T. (1995) Science 268, 825-826.
- Watson, K.L., Justice, R.W. & Bryant, P.J. (1994) J. Cell Sci. 107 Suppl. 18:19-33.
- Martin, C.H., Mayeda, C.A., Davis, C.A., Ericsson, C.L., Knafels, J.D., Mathog, D., Celniker, S., Lewis, E. B., & Palazzolo, M.J.,. (1995) Proc. Natl. Acad. Sci. USA 92, 8398-8402.
- O'Kane, K. & Gehring, W.R. (1987) Proc. Nat. Acad. Sci., USA 84, 9123-9127.
- Kleckner, N., Roth, J. & Botstein, D. (1975) J. Mol. Biol. 116, 125-.
- Bingham, P.M., Levis, R. & Rubin, G.M. (1982) Cell 25, 693-704.
- Spradling, A.C. & Rubin, G.M. (1982) Science 218, 341-347.
- Engels, W.R. (1984) Science 226, 1194-1196.
- Kidwell, M.G. (1987) Dros. Inform. Service 66, 81-83.
- Rubin, G.M. & Spradling, A.C. (1982) Science 218, 348-353.
- Cooley, L., Kelley, R. & Spradling, A.C. (1988) Science 239, 1121- 1128.
- Stellar, H. & Pirrotta, V. (1985) EMBO J. 4, 167-171.
- Brand, A. & Perrimon, N. (1993) Development (Cambridge, U.K.) 118: 401-415.
- Golic, K. & Lindquist, S. (1989) Cell 59, 499-509.
- Bier, E., Vaessin, H., Shepherd, S., Lee, K., McCall, K., Barbel, S., Ackerman, L., Carreto, R., Uemura, T., Grell, E., Jan, L.Y. & Jan, Y. N. (1989) Genes Dev. 3, 1273-1287.
- Karpen, G.H. & Spradling, A.C. (1992) Genetics 132, 737-753.
- Gaul, U., Mardon, G. & Rubin, G.M. (1992) Cell 68, 1007-1019.
- Török, T., Tick, G., Alvarado, M., & Kiss, I. (1993) Genetics 135, 71-80.
- Chang, Z., Price, B.D., Bockheim, S., Boedigheimer, M.J., Smith, R. & Laughon, A. (1993). Devel. Biol. 160, 315-332.
- Cutforth, T. & Rubin, G.M. (1994) Cell 77, 1027-1036.
- FlyBase (1994) Nucleic Acids Res. 22, 3456-3458.
- Tower, J., Karpen, G.H., Craig, N. & Spradling, A.C. (1993) Genetics 133, 347-359.
- Ashburner, A., Thompson, P., Roote, J., Lasko, P.Ï., Grau Y., El Messal, M., Roth, S. & Simpson, P. (1990) Genetics 126, 679-694.
- Kidwell, M.G. (1986) in Drosophila, A Practical Approach ed. Roberts, D.B. (IRL, Oxford) pp 59-81.
- Smith, D., Wohlgemuth, J., Calvi, B.R., Franklin, I. & Gelbart, W.M. (1993) Genetics 135, 1063-76.
- Franz, G., Loukeris, T.G., Dialektaki, G., Thompson, C.R.L., & Savakis, C. (1994). Proc. Nat. Acad. Sci., USA 91, 4746-4750.).
- Tsubota, S., Ashburner, M., & Schedl, P. (1985) Mol. Cell. Biol. 5, 2567-2574.
- Kelley, M. B., Kidd, S., Berg, R.L. & Young, M.W. (1987) Mol. Cell. Biol. 7, 1545-1548.
- Sandmeyer, S.B., Hansen, L.J. , & Chalder, D.L. (1990) Annu. Rev. Genet. 24, 491-518.
- Liebman, S.W. & Newnam, G. (1993) Genetics 133, 499-510.
- Kirchner, J., Connolly, C.M.& Sandmeyer, S.B. (1995) Science 267, 1488-1491.
- Bossy, B.L.M., Hall, C. & Spierer P. (1983). EMBO J. 3, 2537-2541.
- Kozlova, A., Semeshin, V.F., Tretyakova, I.V., Kokoza, E.B., Pirrotta, V, Grafodatskaya, V.E., Belyaeva, E.S. & Zhimulev, I.F. (1994) Genetics 136, 1063-1073.
- Dujon, B., Alexandraki, D., Andre, B., Ansorge, W., Baladron, V., et al. (1994) Nature (London) 369, 371-378.
- Sulston, J., Du, Z., Thomas, K., Wilson, R., Hillier, L., Staden, R., Halloran, N., Green, P., Thierry-Mieg, J., Qiu, L., Dear, S., Coulson, A., Craxton, M., Durbin, R., Berks, M., Metzstein, M., Hawkins, T., Ainscough, R. & Waterston, R. (1992) Nature (London) 356, 37-41.
- Konopka, R.J. & Benzer, S. (1971) Proc. Natl. Acad. Sci. USA 68, 2112-2116.